This function, part of the MicrobiomeStat package, generates various types of plots to visualize alpha diversity measures in microbiome data. It supports different visualization schemes for single time point, paired time points, and longitudinal data.
Usage
plot_alpha_diversity(
alpha.obj,
meta.dat,
measure,
group.var = NULL,
strata.var = NULL,
subject.var = NULL,
time.var = NULL,
adj.vars = NULL,
time.point.plot,
is.plot.change = FALSE,
alpha.change.func = "log fold change",
plot.type = c("boxplot", "spaghettiplot"),
...
)Arguments
- alpha.obj
A required list of pre-calculated alpha diversity indices, typically returned by `mStat_calculate_alpha_diversity()`. This legacy wrapper does not accept a raw feature table and therefore cannot calculate alpha diversity internally.
- meta.dat
A data frame containing metadata for the samples.
- measure
The alpha diversity index to be plotted. Supported indices include "shannon", "simpson", "observed_species", "chao1", "ace", "pielou", and "faith_pd".
- group.var
Character string specifying the grouping variable in meta.dat.
- strata.var
Character string specifying the stratification variable in meta.dat.
- subject.var
Character string specifying the subject variable in meta.dat.
- time.var
Character string specifying the time variable in meta.dat.
- adj.vars
Character vector specifying variables to adjust for. If specified, residuals will be used.
- time.point.plot
Character vector specifying time points to plot. The first point will be the reference.
- is.plot.change
Logical, whether to plot change from baseline. Only effective when there are multiple time points.
- alpha.change.func
Character string specifying the function to calculate alpha diversity change, default is "log fold change".
- plot.type
Character string specifying the plot type, one of c("boxplot", "spaghettiplot").
- ...
Additional arguments passed to the underlying plotting functions.
Details
As a core component of the MicrobiomeStat package, this function leverages the package's data structures and conventions to provide flexible and powerful visualizations of alpha diversity in microbiome studies. It integrates seamlessly with other MicrobiomeStat functions for comprehensive microbiome data analysis.
This function provides a flexible framework for visualizing alpha diversity in microbiome data. It can handle single time point, paired time points, and longitudinal data. The function offers different plot types including box plots and spaghetti plots, and can visualize changes over time.
This function provides a flexible framework for visualizing alpha diversity in microbiome data. It can handle single time point, paired time points, and longitudinal data. The function offers different plot types including box plots and spaghetti plots, and can visualize changes over time.
The behavior of the function depends on the number of time points specified: - For a single time point or when time variable is not specified, only boxplot is available. - For two time points, both boxplot and spaghettiplot are available, with an option to plot change. - For more than two time points, both boxplot and spaghettiplot are available for raw measures, but change plots are not supported.
When `is.plot.change` is TRUE and multiple time points are specified, the function will calculate changes from the baseline (first time point) using the method specified in `alpha.change.func`.
Examples
data(peerj32.obj)
alpha.obj <- mStat_calculate_alpha_diversity(peerj32.obj$feature.tab,
alpha.name = c("shannon", "observed_species"))
#> Warning: It appears the data may not have been rarefied. Please verify.
#> Calculating shannon diversity...
#> Calculating observed_species diversity...
#> Diversity calculations complete.
plot_alpha_diversity(alpha.obj = alpha.obj,
meta.dat = peerj32.obj$meta.dat,
measure = c("shannon", "observed_species"),
group.var = "group",
strata.var = "sex",
time.var = "time",
time.point.plot = c("1"),
plot.type = "boxplot")
#> Validation passed.
#> Data has been subsetted based on the provided samIDs.
#> Updated metadata to match the subsetted data.
#> 22 samples were excluded.
#> Updated feature table to match the subsetted data.
#> Pruned all-zero features after subsetting.
#> Data subsetting complete. Returning updated data object.
#> $shannon
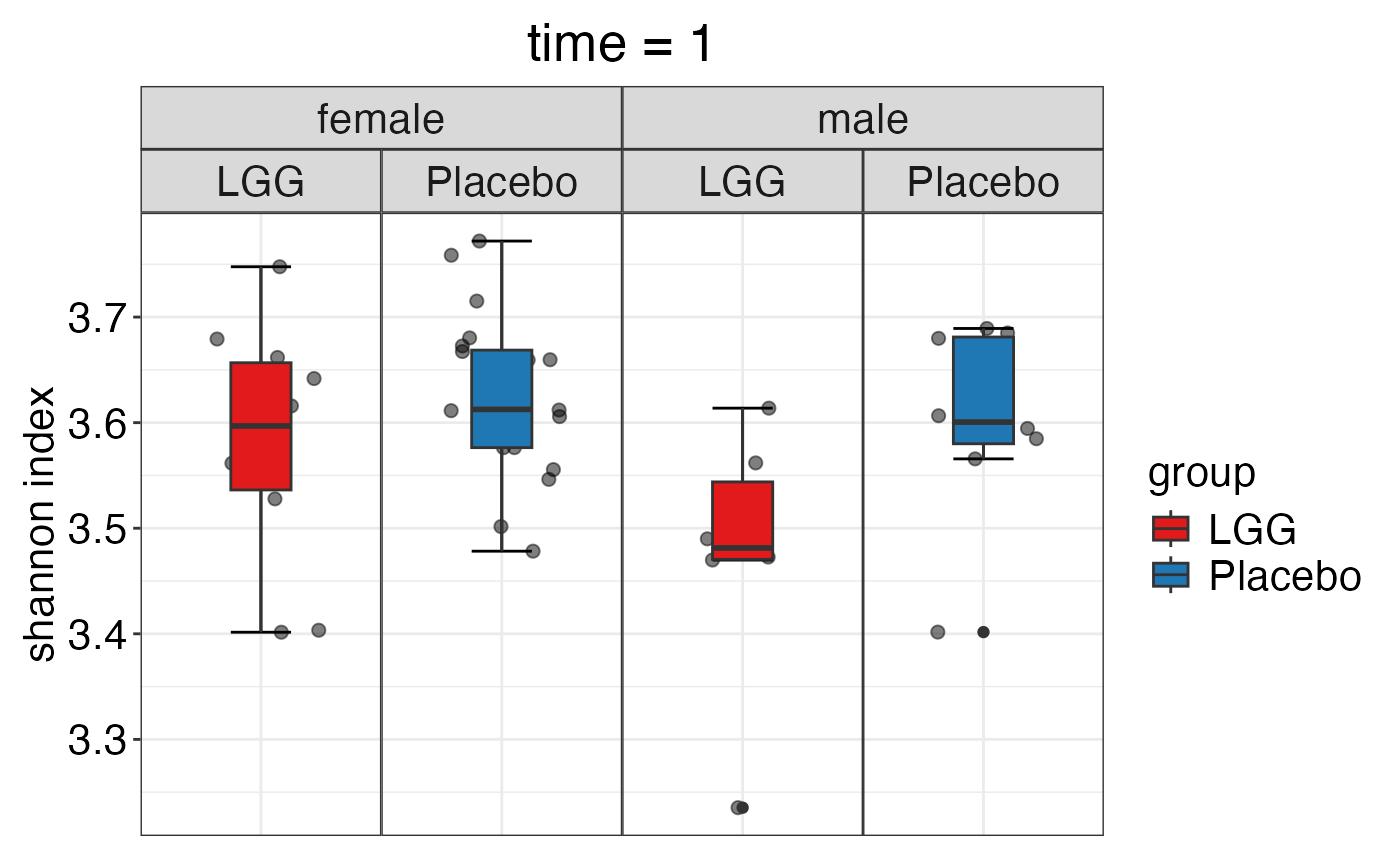 #>
#> $observed_species
#>
#> $observed_species
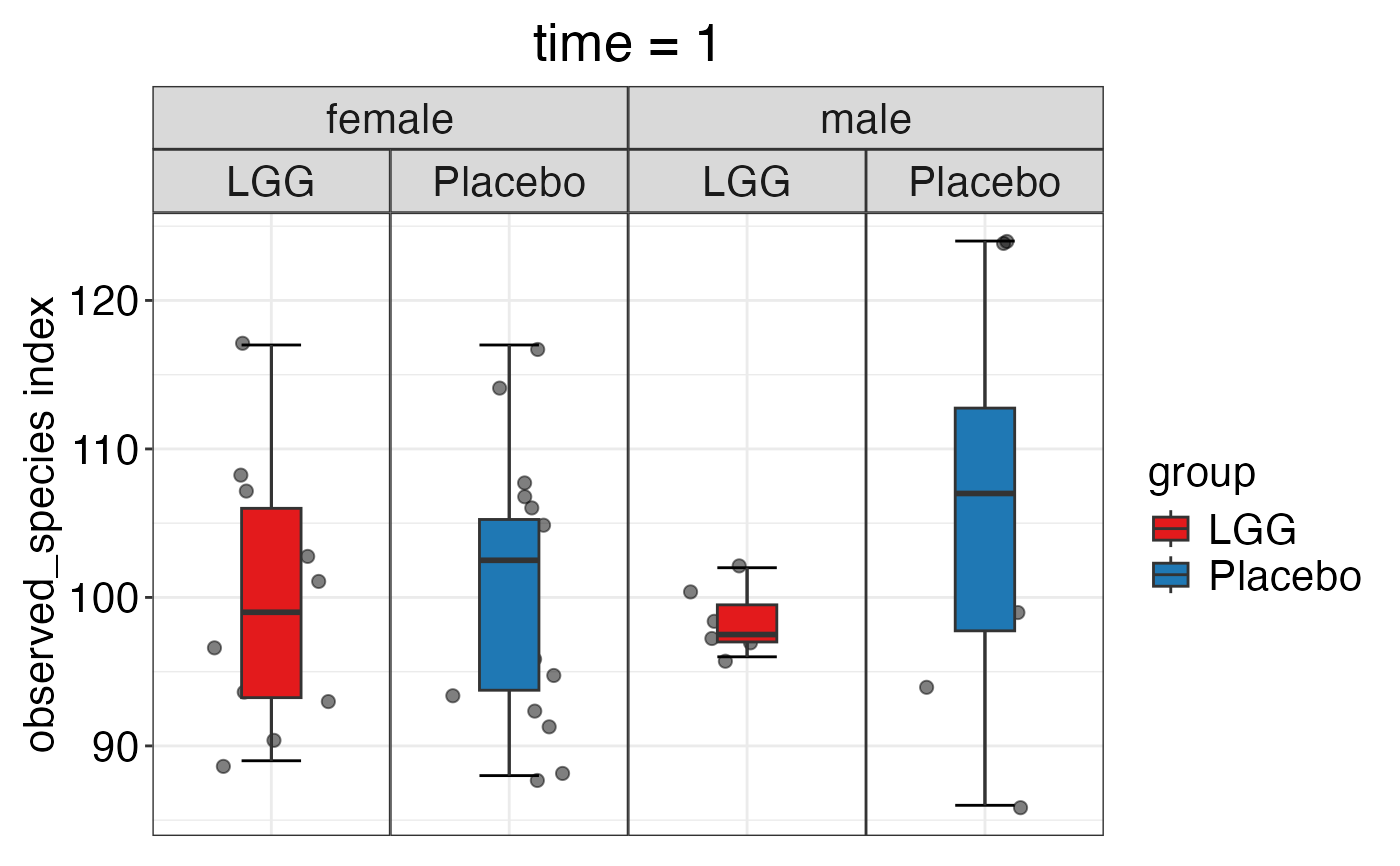 #>
plot_alpha_diversity(alpha.obj = alpha.obj,
meta.dat = peerj32.obj$meta.dat,
measure = c("shannon", "observed_species"),
group.var = "group",
subject.var = "subject",
strata.var = "sex",
time.var = "time",
time.point.plot = c("1", "2"),
is.plot.change = TRUE,
plot.type = "boxplot")
#> $shannon
#>
plot_alpha_diversity(alpha.obj = alpha.obj,
meta.dat = peerj32.obj$meta.dat,
measure = c("shannon", "observed_species"),
group.var = "group",
subject.var = "subject",
strata.var = "sex",
time.var = "time",
time.point.plot = c("1", "2"),
is.plot.change = TRUE,
plot.type = "boxplot")
#> $shannon
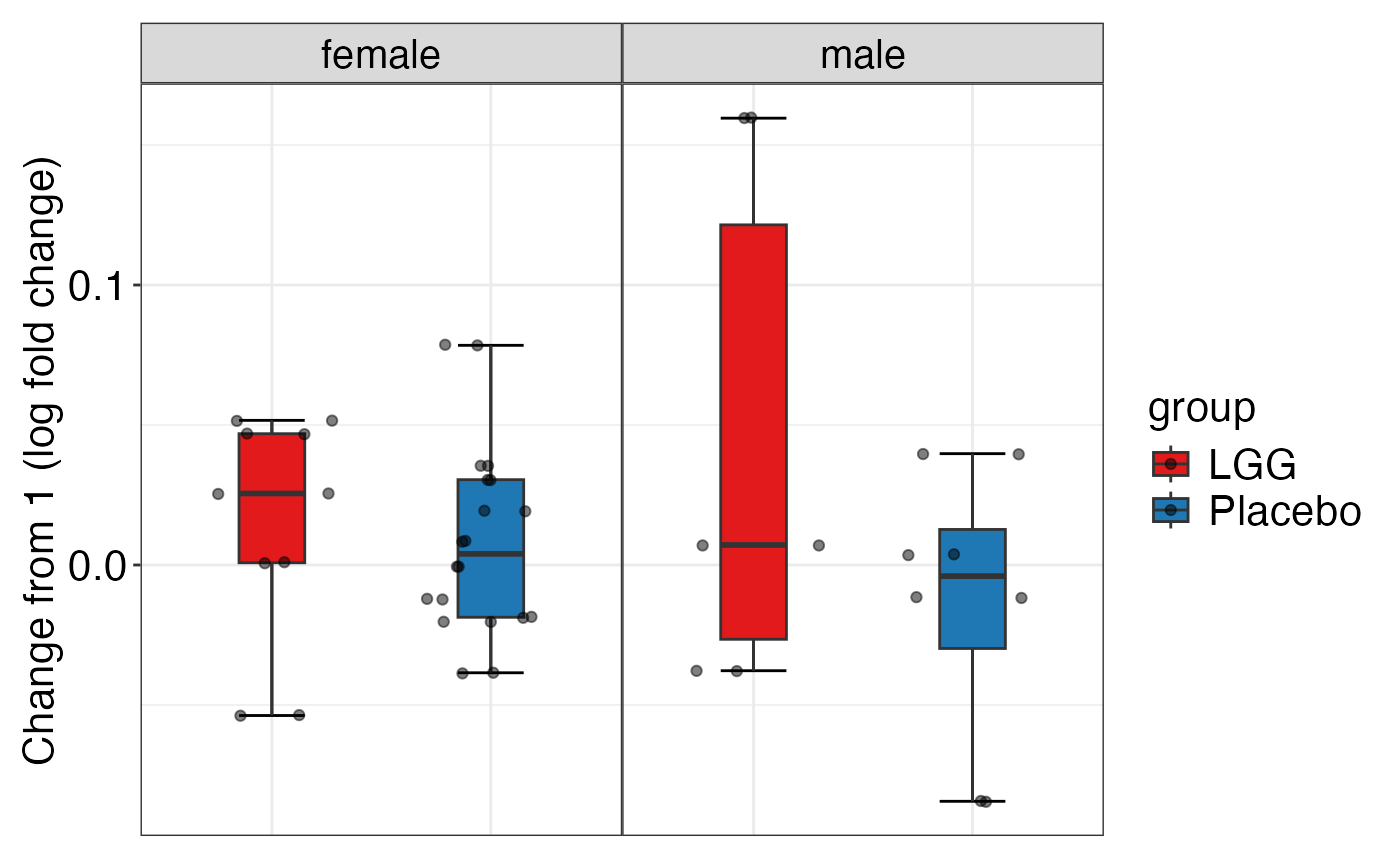 #>
#> $observed_species
#>
#> $observed_species
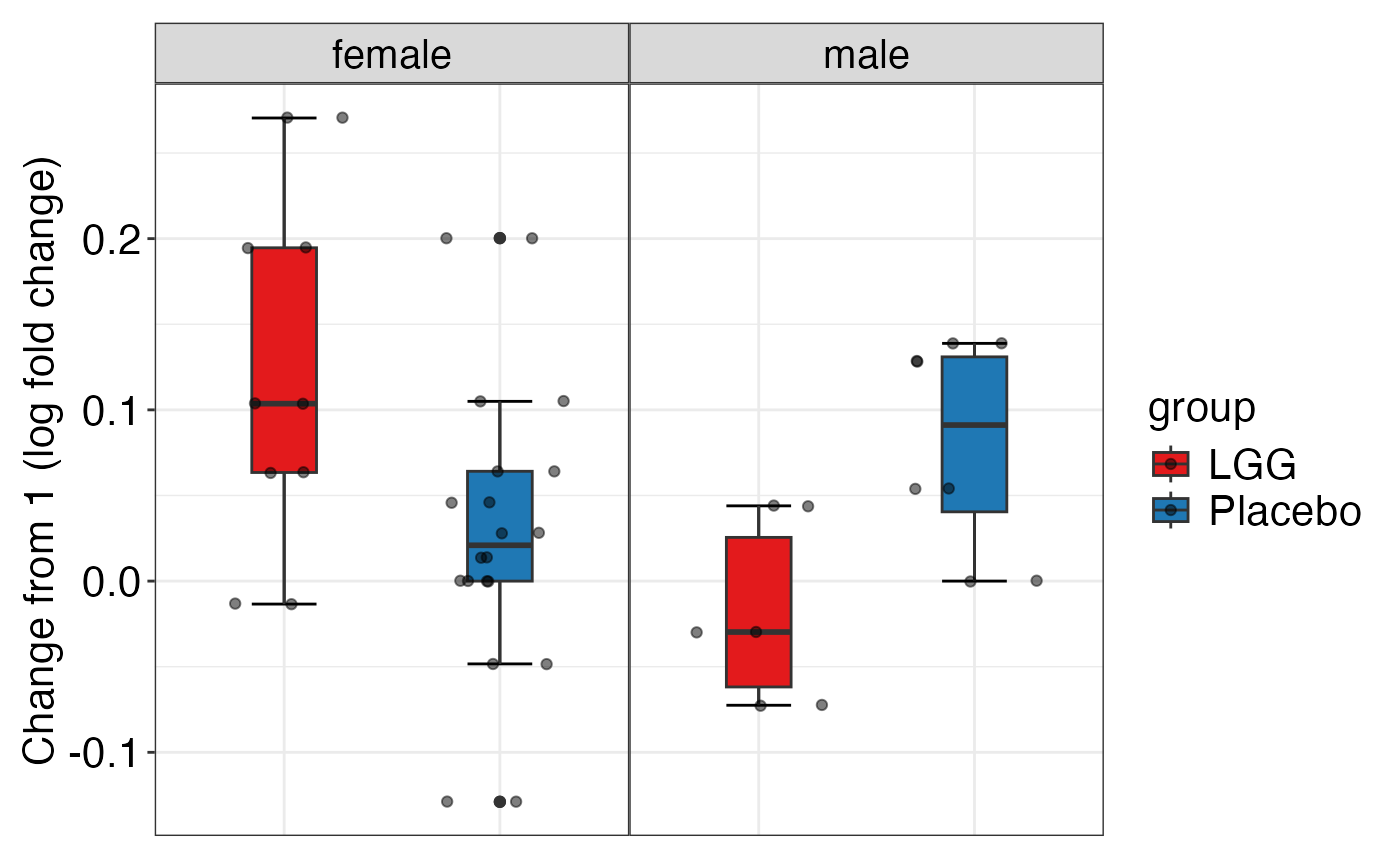 #>
plot_alpha_diversity(alpha.obj = alpha.obj,
meta.dat = peerj32.obj$meta.dat,
measure = c("shannon", "observed_species"),
group.var = "group",
subject.var = "subject",
strata.var = "sex",
time.var = "time",
time.point.plot = c("1", "2"),
is.plot.change = FALSE,
plot.type = "boxplot")
#> $shannon
#>
plot_alpha_diversity(alpha.obj = alpha.obj,
meta.dat = peerj32.obj$meta.dat,
measure = c("shannon", "observed_species"),
group.var = "group",
subject.var = "subject",
strata.var = "sex",
time.var = "time",
time.point.plot = c("1", "2"),
is.plot.change = FALSE,
plot.type = "boxplot")
#> $shannon
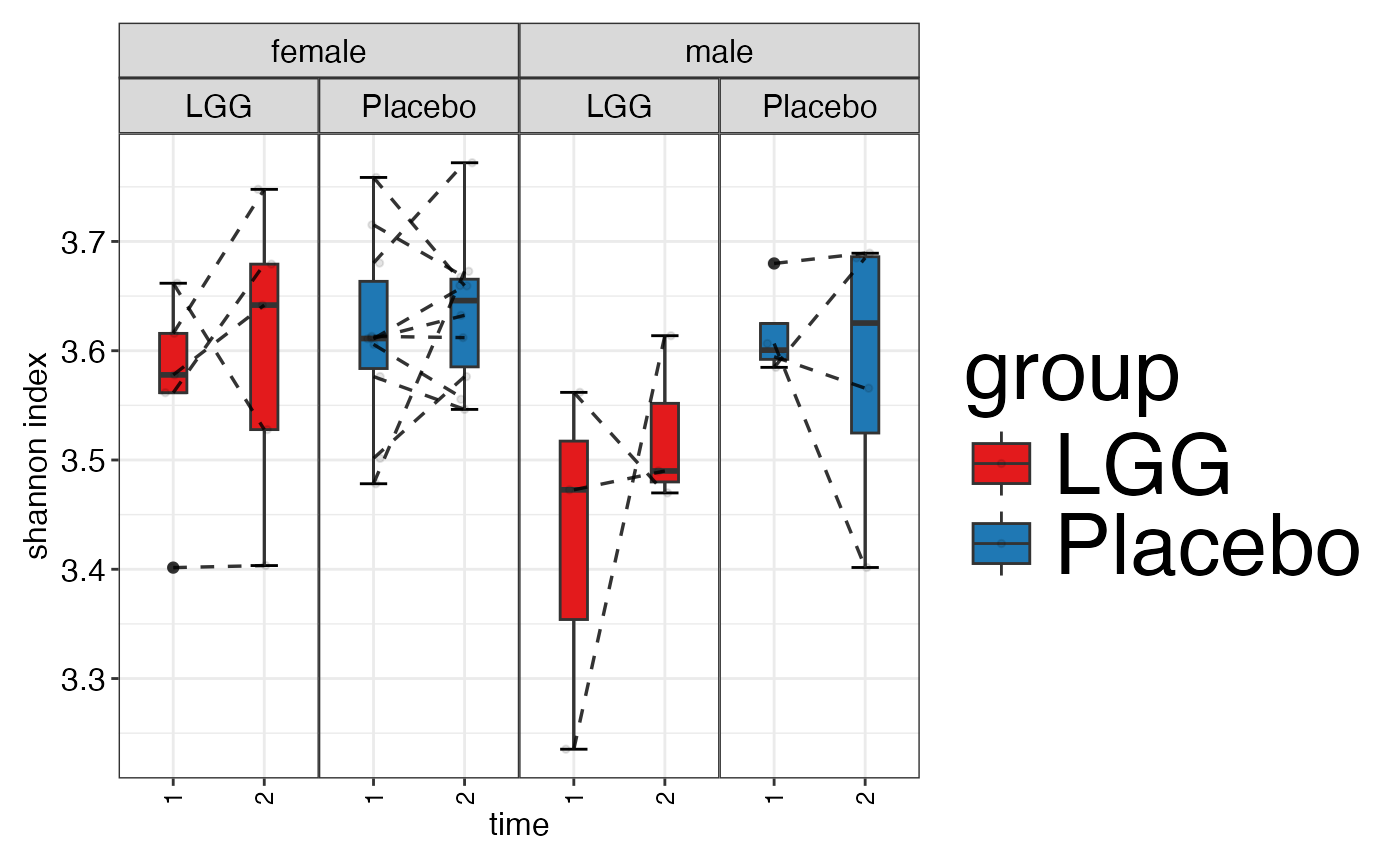 #>
#> $observed_species
#>
#> $observed_species
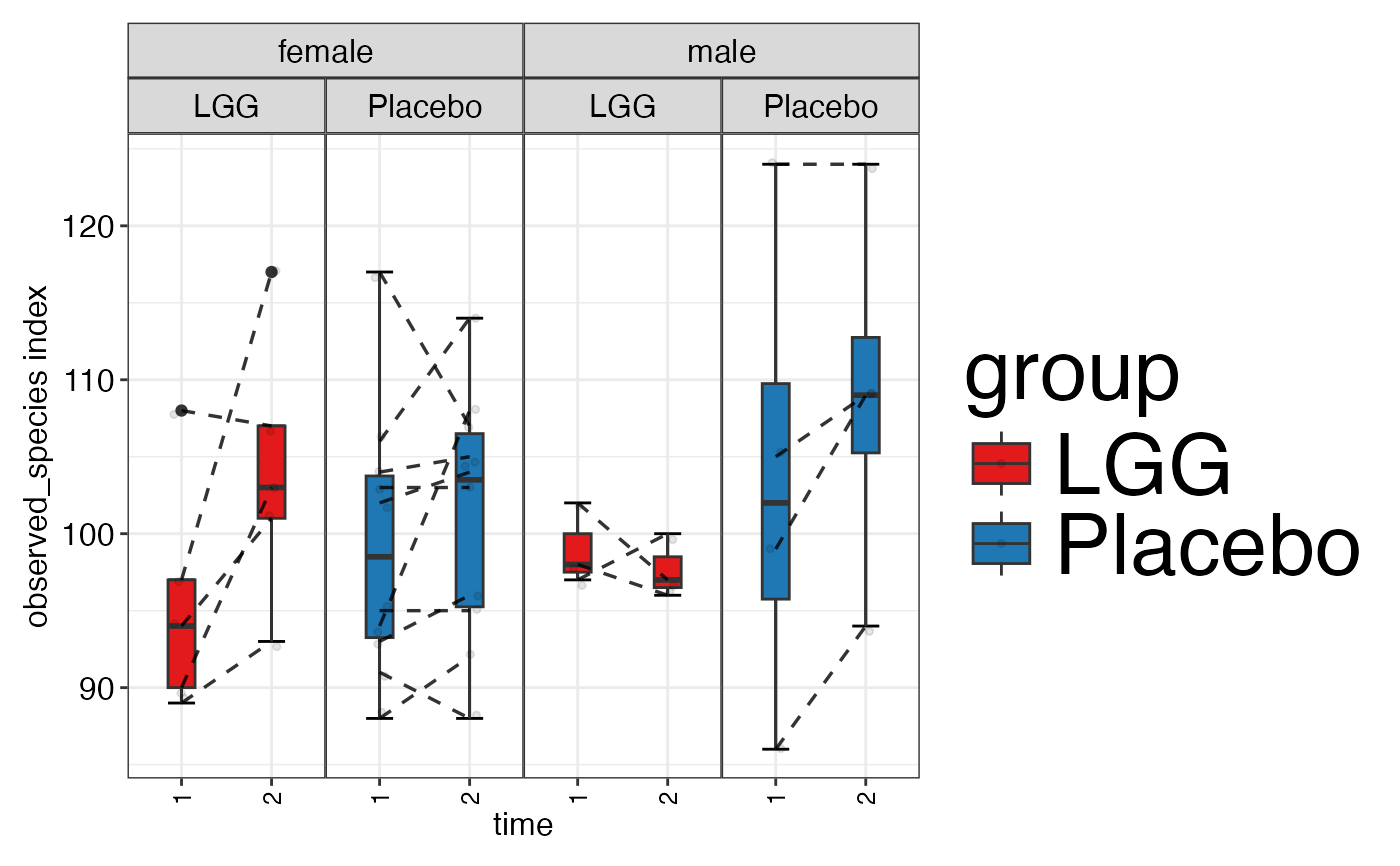 #>
plot_alpha_diversity(alpha.obj = alpha.obj,
meta.dat = peerj32.obj$meta.dat,
measure = c("shannon", "observed_species"),
group.var = "group",
subject.var = "subject",
strata.var = "sex",
time.var = "time",
time.point.plot = c("1", "2"),
is.plot.change = FALSE,
plot.type = "spaghettiplot")
#> Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
#> ℹ Please use `linewidth` instead.
#> ℹ The deprecated feature was likely used in the MicrobiomeStat package.
#> Please report the issue at
#> <https://github.com/cafferychen777/MicrobiomeStat/issues>.
#> $shannon
#>
plot_alpha_diversity(alpha.obj = alpha.obj,
meta.dat = peerj32.obj$meta.dat,
measure = c("shannon", "observed_species"),
group.var = "group",
subject.var = "subject",
strata.var = "sex",
time.var = "time",
time.point.plot = c("1", "2"),
is.plot.change = FALSE,
plot.type = "spaghettiplot")
#> Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
#> ℹ Please use `linewidth` instead.
#> ℹ The deprecated feature was likely used in the MicrobiomeStat package.
#> Please report the issue at
#> <https://github.com/cafferychen777/MicrobiomeStat/issues>.
#> $shannon
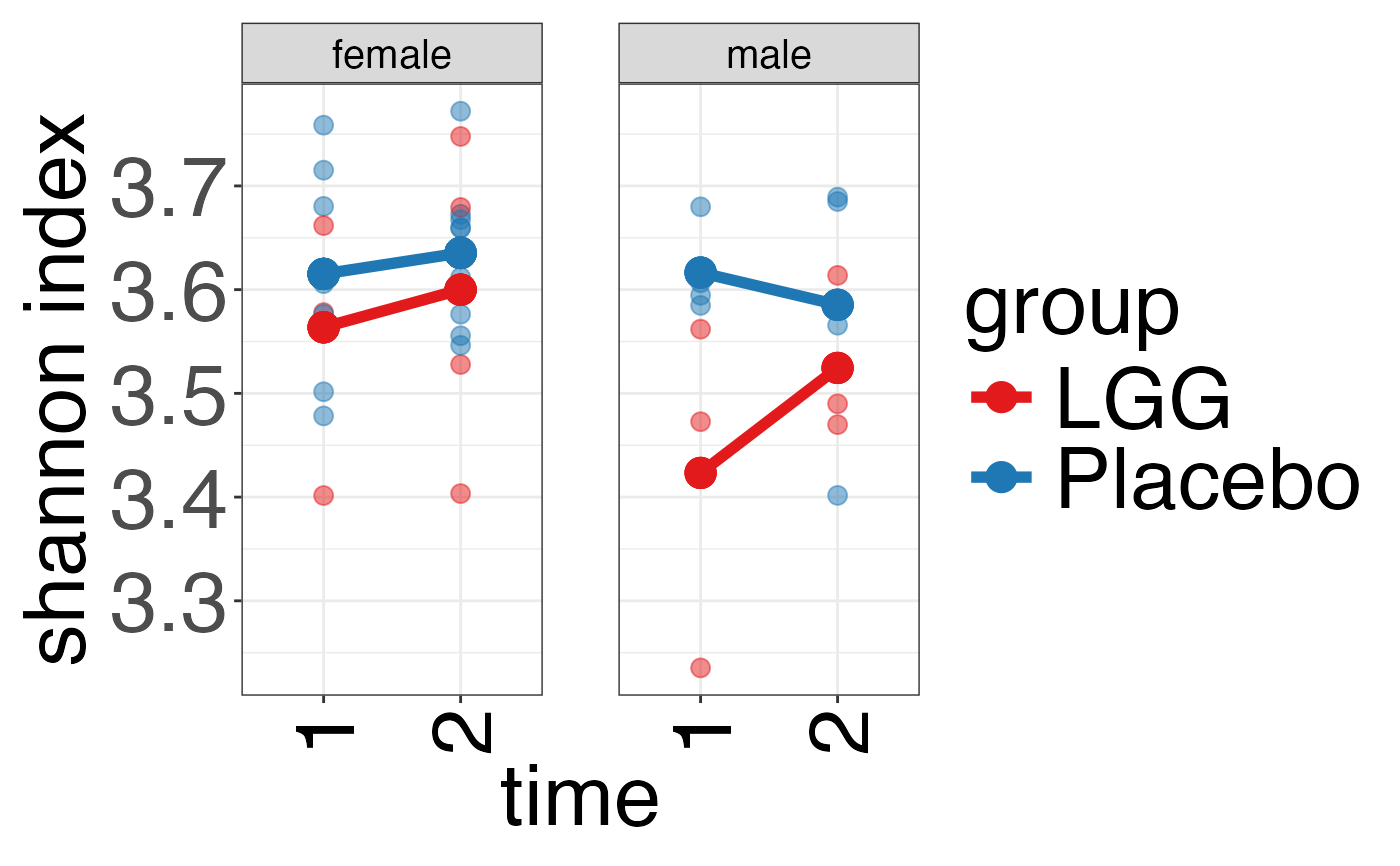 #>
#> $observed_species
#>
#> $observed_species
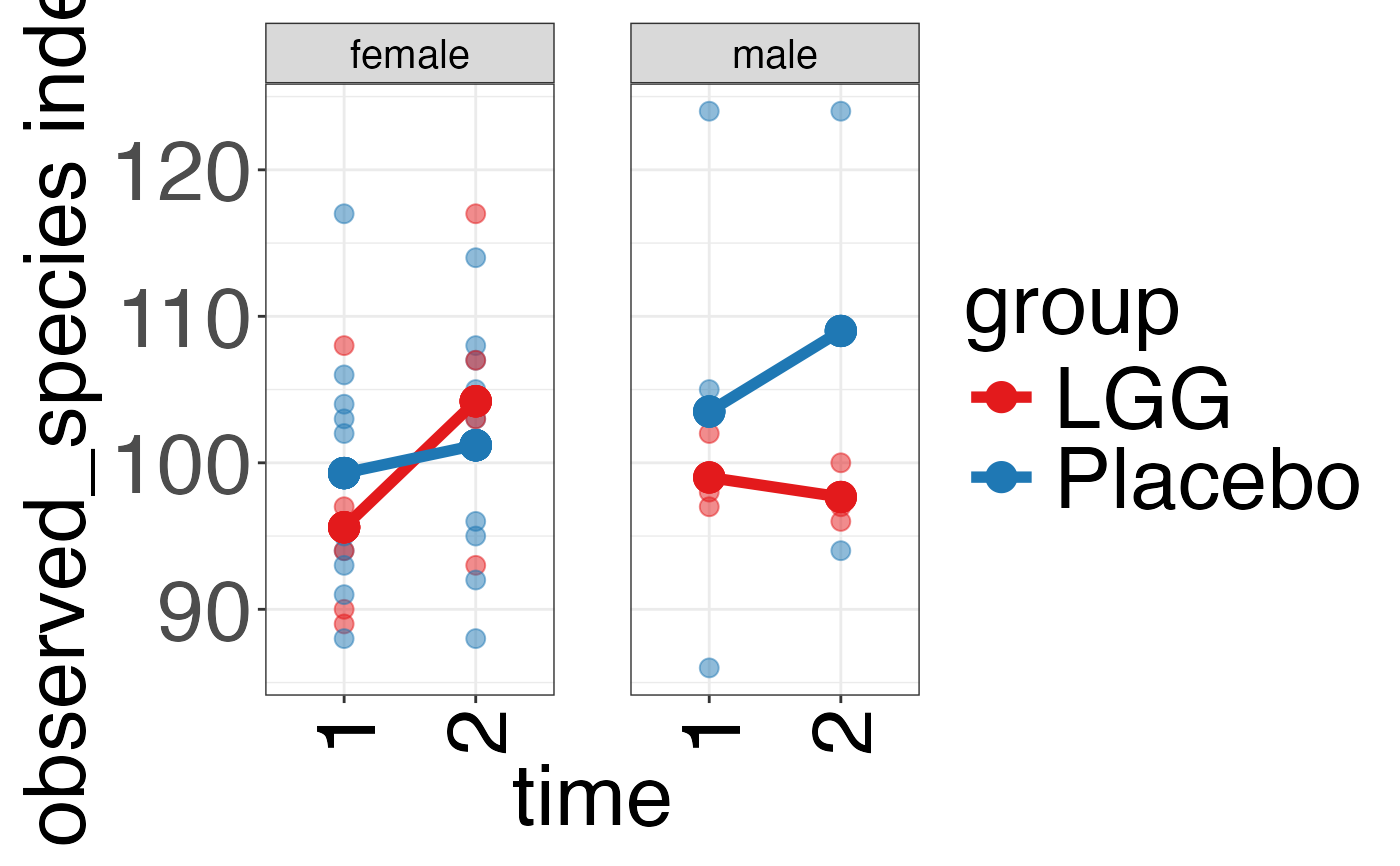 #>
data(ecam.obj)
alpha.obj <- mStat_calculate_alpha_diversity(ecam.obj$feature.tab,
alpha.name = c("shannon", "observed_species"))
#> Warning: It appears the data may not have been rarefied. Please verify.
#> Calculating shannon diversity...
#> Calculating observed_species diversity...
#> Diversity calculations complete.
plot_alpha_diversity(alpha.obj = alpha.obj,
meta.dat = ecam.obj$meta.dat,
measure = c("shannon", "observed_species"),
group.var = "delivery",
subject.var = "antiexposedall",
strata.var = "diet",
time.var = "month_num",
time.point.plot = unique(ecam.obj$meta.dat$month_num),
is.plot.change = FALSE,
plot.type = "spaghettiplot")
#> $shannon
#>
data(ecam.obj)
alpha.obj <- mStat_calculate_alpha_diversity(ecam.obj$feature.tab,
alpha.name = c("shannon", "observed_species"))
#> Warning: It appears the data may not have been rarefied. Please verify.
#> Calculating shannon diversity...
#> Calculating observed_species diversity...
#> Diversity calculations complete.
plot_alpha_diversity(alpha.obj = alpha.obj,
meta.dat = ecam.obj$meta.dat,
measure = c("shannon", "observed_species"),
group.var = "delivery",
subject.var = "antiexposedall",
strata.var = "diet",
time.var = "month_num",
time.point.plot = unique(ecam.obj$meta.dat$month_num),
is.plot.change = FALSE,
plot.type = "spaghettiplot")
#> $shannon
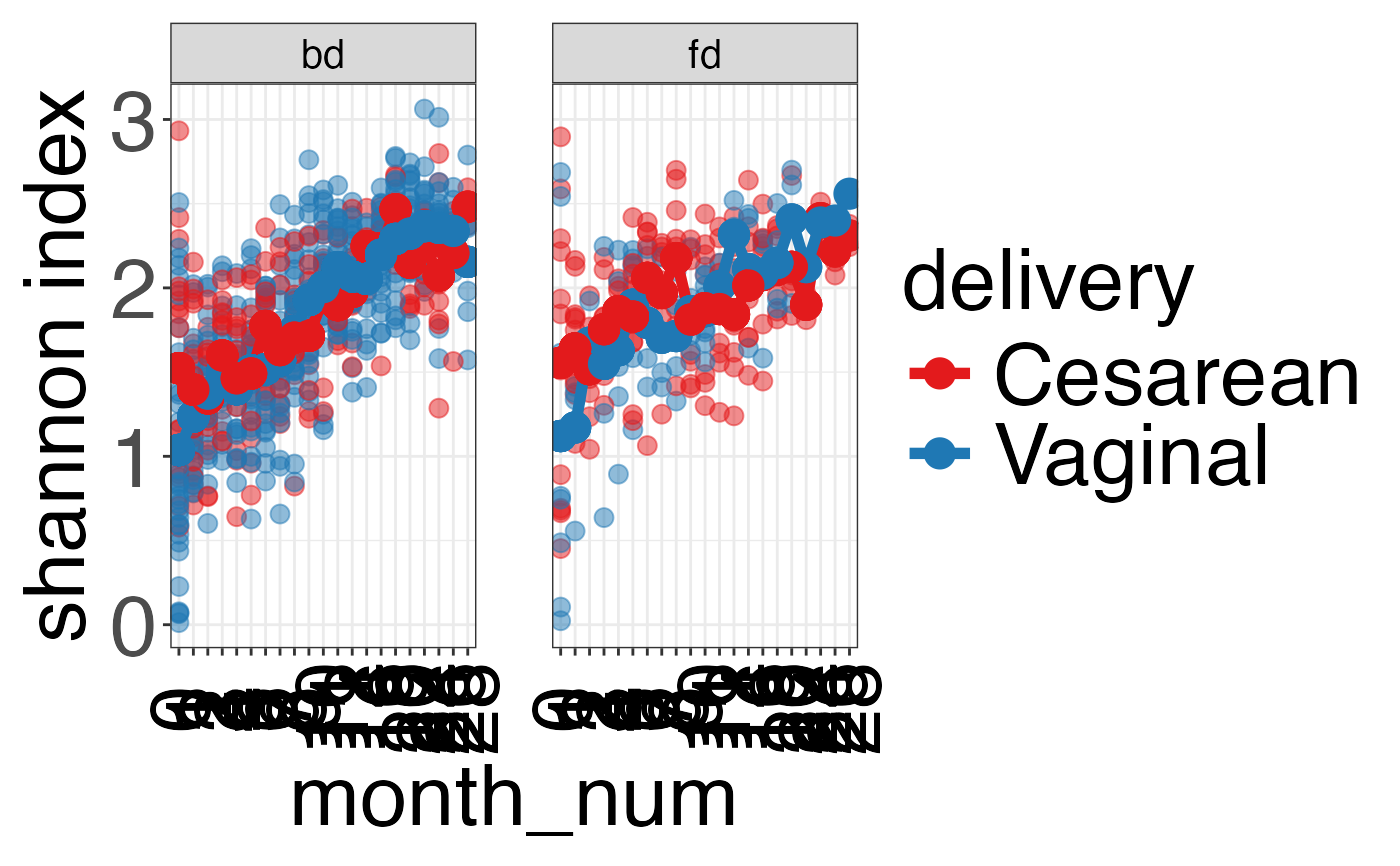 #>
#> $observed_species
#>
#> $observed_species
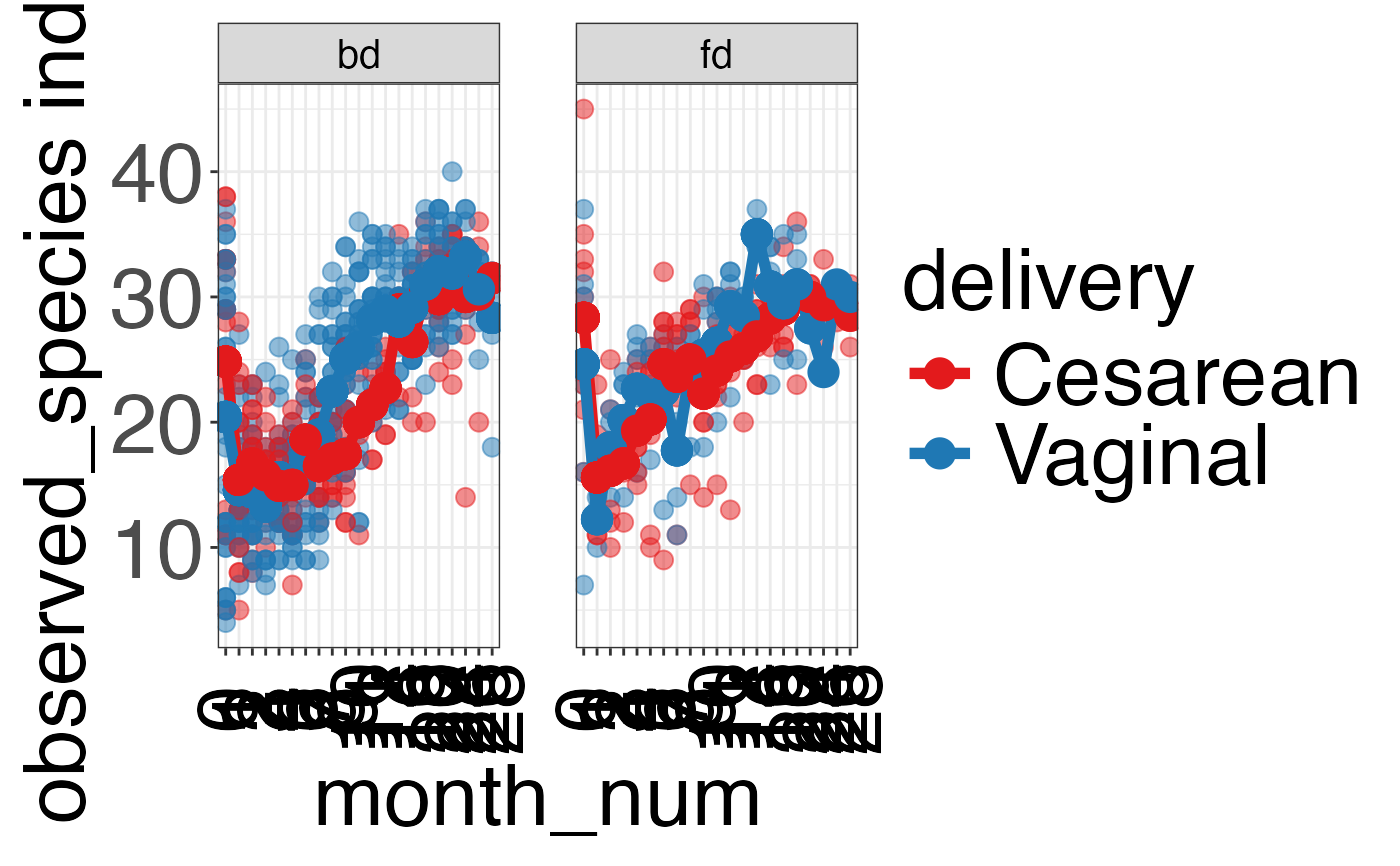 #>
plot_alpha_diversity(alpha.obj = alpha.obj,
meta.dat = ecam.obj$meta.dat,
measure = c("shannon", "observed_species"),
group.var = "delivery",
subject.var = "antiexposedall",
strata.var = "diet",
time.var = "month_num",
time.point.plot = unique(ecam.obj$meta.dat$month_num),
is.plot.change = FALSE,
plot.type = "boxplot")
#> $shannon
#>
plot_alpha_diversity(alpha.obj = alpha.obj,
meta.dat = ecam.obj$meta.dat,
measure = c("shannon", "observed_species"),
group.var = "delivery",
subject.var = "antiexposedall",
strata.var = "diet",
time.var = "month_num",
time.point.plot = unique(ecam.obj$meta.dat$month_num),
is.plot.change = FALSE,
plot.type = "boxplot")
#> $shannon
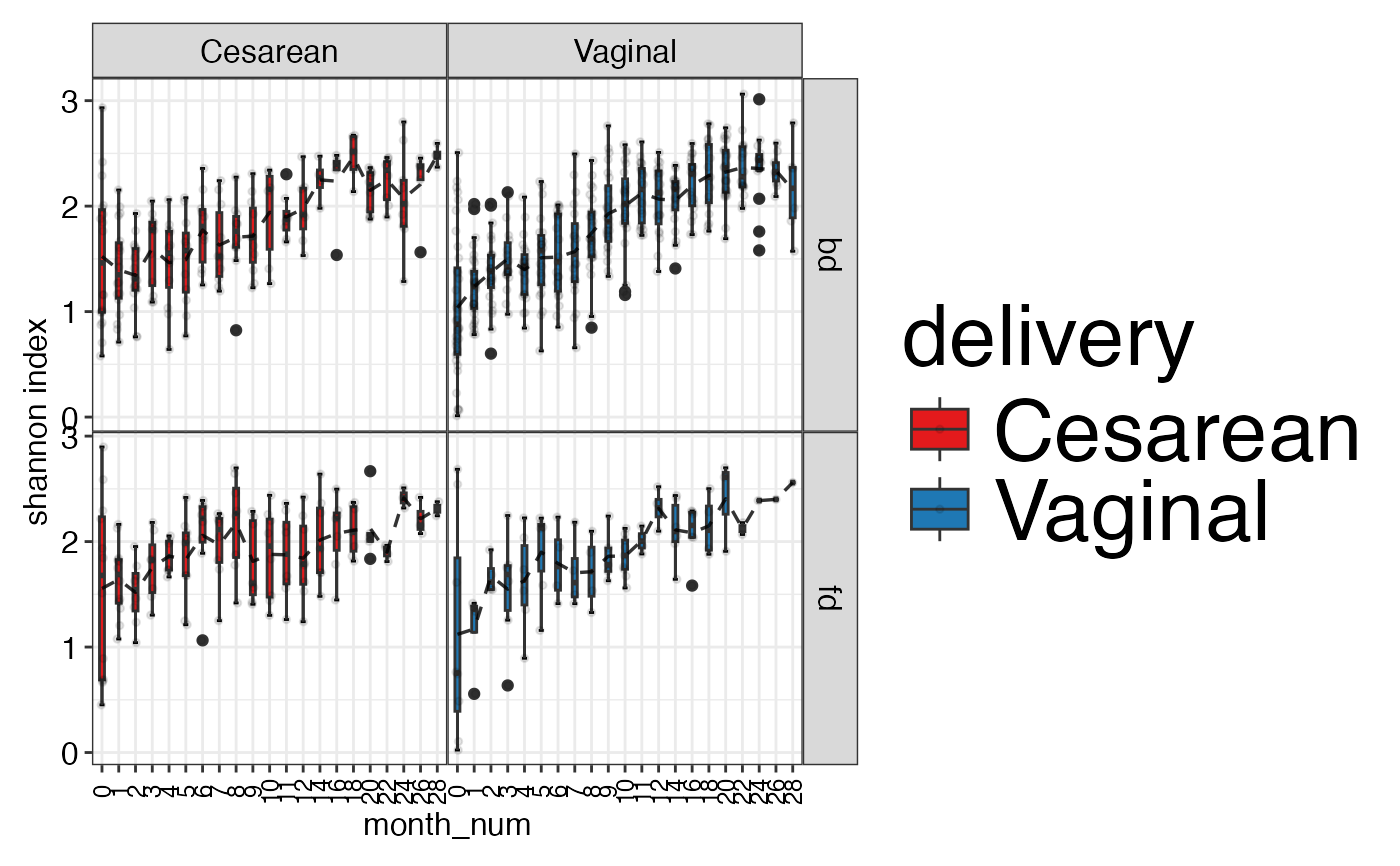 #>
#> $observed_species
#>
#> $observed_species
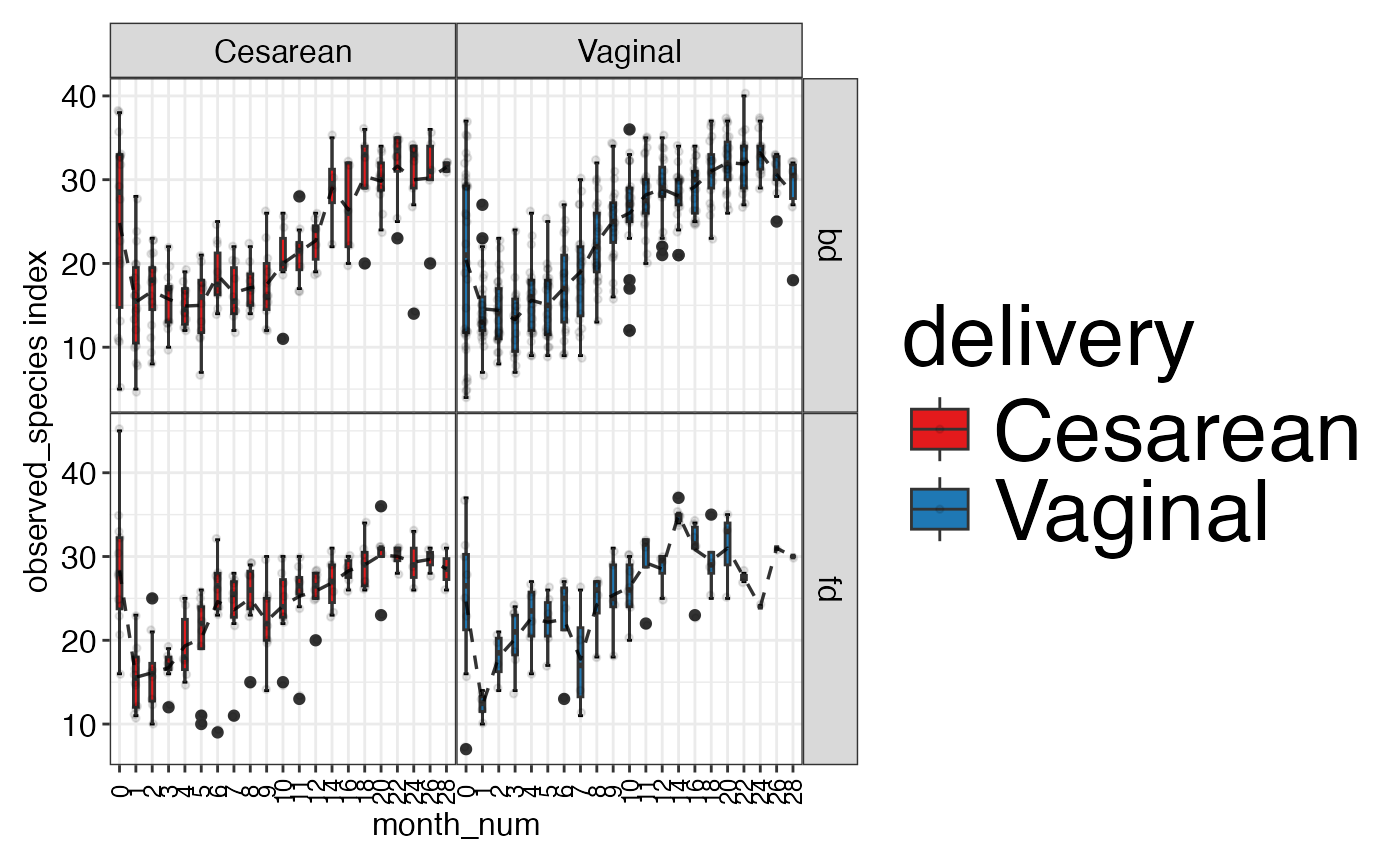 #>
#>